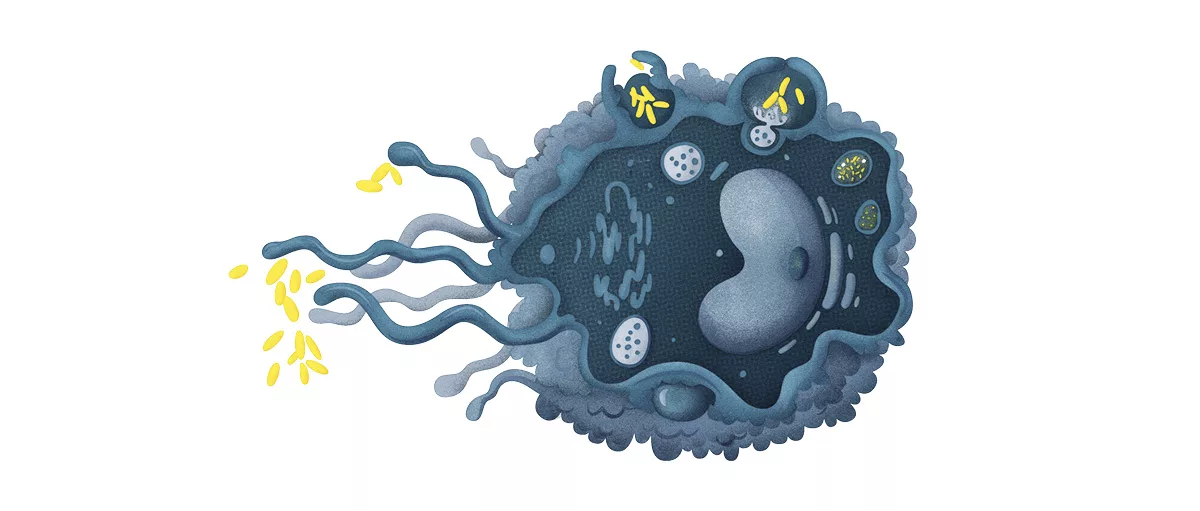
Imagine your body as a bustling city. Within that city, macrophages are the night‑watch officers—always on patrol, ready to call the alarm, clean up messes, or calm a crowd. When their communication lines—macrophage pathways—work smoothly, everything runs peacefully. When those pathways go haywire, they can spark inflammation, damage joints, or even fuel chronic lung disease. In the next few minutes we’ll walk through the main signaling routes, see how they turn up the heat in common ailments, and explore what the latest science is doing to tame them. Ready? Let’s dive in together.
Core Signaling Routes
What are the canonical macrophage signaling cascades?
Macrophages speak using a handful of well‑studied highways. The most frequent ones you’ll hear about are:
- Toll‑like receptor (TLR) → NF‑κB – the classic “danger alarm” route.
- JAK/STAT – the cytokine‑relay system.
- MAPK (ERK, p38, JNK) – the stress‑response network.
- PI3K‑Akt / mTOR – the metabolic switch that decides whether the cell burns fuel fast or slow.
- NLRP3 inflammasome – the “boiling pot” for IL‑1β and IL‑18.
- cGAS‑STING – the DNA‑damage detector.
Each pathway can operate alone or in concert with the others, forming a mesh of checks and balances that determines whether a macrophage will become pro‑inflammatory, anti‑inflammatory, or stay in a neutral, tissue‑repair mode.
Key pathways at a glance
Below is a quick cheat‑sheet you can bookmark. Think of it as a “macrophage GPS” guide.
| Pathway | Primary Trigger | Major Outcome |
|---|---|---|
| TLR/NF‑κB | LPS, bacterial DNA, viral RNA | Rapid production of TNF‑α, IL‑6, IL‑1β |
| JAK/STAT | Cytokines (IFN‑γ, IL‑6, IL‑10) | Gene‑programming for activation or suppression |
| MAPK | Stress, ROS, cytokines | Cellular proliferation, apoptosis, cytokine release |
| PI3K‑Akt/mTOR | Growth factors, nutrients | Metabolic re‑wiring; favors M2‑like repair phenotype |
| NLRP3 Inflammasome | Crystals, ATP, ROS | IL‑1β & IL‑18 maturation → fever, pain |
| cGAS‑STING | Cytosolic DNA (viral, mitochondrial) | Type‑I interferon response |
How does Toll‑like receptor (TLR) activation drive NF‑κB in macrophages?
Picture a fire alarm. When a TLR (think TLR4 for bacterial LPS) detects a threat, it recruits the adaptor protein MyD88. MyD88 then calls in IRAK kinases, which hand the signal to the IKK complex. IKK phosphorylates IκB, the “gate‑keeper” that normally holds NF‑κB in the cytoplasm. Once IκB is shredded, NF‑κB rushes into the nucleus and flips on genes for TNF‑α, IL‑6, and many other inflammatory mediators.
Why the fuss? Because an over‑zealous NF‑κB response is what turns a mild infection into a septic shock, and it also fuels chronic joints pain in rheumatoid arthritis and osteoarthritis.
Step‑by‑step flow
- Ligand (e.g., LPS) binds TLR4 on the macrophage surface.
- MyD88 adaptor is recruited.
- IRAK4/1 activate the TRAF6 complex.
- IKKβ phosphorylates IκBα.
- IκBα degrades, freeing NF‑κB (p65/p50).
- NF‑κB enters nucleus → transcription of pro‑inflammatory genes.
When does the JAK/STAT axis become pathogenic?
JAK/STAT is the “mail‑room” for cytokine messages. A cytokine binds its receptor, JAKs attached to the receptor get phosphorylated, and then they tag STAT proteins. Phosphorylated STATs dimerize, slide into the nucleus, and rewrite the cell’s instruction manual.
In a healthy setting, STAT1 drives a classic “M1” (pro‑inflammatory) program in response to IFN‑γ, while STAT3 or STAT6 leans the cell toward an “M2” (repair‑focused) stance when IL‑10 or IL‑4 are present. Problems arise when the balance tips. Persistent IL‑6 signaling, for instance, forces chronic STAT3 activation—one of the hallmarks of inflammatory joint diseases such as rheumatoid arthritis.
Statistical snapshot
According to a 2023 review in Nature Reviews Immunology, patients with high serum IL‑6 have a 2.4‑fold increased risk of developing erosive joint damage within five years. The same study also notes that JAK inhibitors (e.g., tofacitinib) reduce that risk by roughly 40%.
What triggers the NLRP3 inflammasome in macrophages?
Think of NLRP3 as a pressure cooker. It needs two cues:
- Signal 1 (priming): often a TLR/NF‑κB event that raises the amount of NLRP3 protein and pro‑IL‑1β.
- Signal 2 (activation): a physical or chemical stressor—crystals (uric acid, calcium pyrophosphate), silica particles, ATP, or mitochondrial ROS—that forces NLRP3 to assemble a complex with ASC and pro‑caspase‑1.
When the complex snaps together, caspase‑1 chops pro‑IL‑1β into its active, fever‑inducing form. This is the molecular engine behind gout flares, gout and CPPD, and silicosis‑related lung inflammation.
Crystal‑driven activation table
| Crystal / Particle | Source | Primary Pathway Triggered |
|---|---|---|
| Uric Acid (Monosodium Urate) | Purine metabolism, diet | NLRP3 inflammasome → IL‑1β |
| Calcium Pyrophosphate (CPP) | Cartilage breakdown | NLRP3 inflammasome → IL‑1β |
| Silica | Occupational inhalation | NLRP3 + NF‑κB → fibrosis |
| Asbestos / Nanotubes | Industrial exposure | ROS + MAPK → chronic inflammation |
How do metabolic cues (PI3K‑Akt, mTOR) rewire macrophage phenotype?
Metabolism is the silent director of immune behavior. When glucose is abundant, PI3K‑Akt ramps up glycolysis, fueling a rapid, “M1‑like” burst of cytokines. In contrast, when fatty acids dominate, mTORC1 activation shifts the cell toward oxidative phosphorylation, encouraging an “M2‑like” tissue‑repair mode.
This metabolic flexibility explains why obesity—where excess nutrients push macrophages into a chronic, low‑grade inflammatory state—exacerbates atherosclerosis, type‑2 diabetes, and even some cancers.
Key metabolic enzymes
Recent work (Cell Metabolism, 2022) highlighted two enzymes that act as switches:
- Hexokinase‑2 (HK2) – promotes glycolysis, favors pro‑inflammatory signaling.
- Isocitrate dehydrogenase‑2 (IDH2) – supports the TCA cycle, nudges cells toward anti‑inflammatory output.
Can cGAS‑STING sensing of cytosolic DNA influence macrophage pathways?
When mitochondria or viruses leak DNA into the cytoplasm, cGAS sniffs it out and synthesizes cGAMP. cGAMP binds STING on the endoplasmic reticulum, sparking a cascade that ends with type‑I interferon production.
While crucial for antiviral defense, chronic activation can drive auto‑immune diseases like systemic lupus erythematosus. Researchers are now testing STING antagonists as potential treatments for such disorders.
Pathways in Diseases
How do macrophage pathways exacerbate inflammatory joint diseases?
In rheumatoid arthritis (RA), synovial macrophages sit at the heart of the storm. Persistent NF‑κB signaling pumps out TNF‑α, IL‑1β, and IL‑6, drawing more immune cells into the joint. Simultaneously, JAK/STAT keeps the fire alive by amplifying the cytokine loop.
Clinically, blocking TNF‑α (with infliximab) or inhibiting JAK (with baricitinib) brings relief for many patients, but about 30% still experience flares—showing we still have work to do on the pathway level.
Why are gout and CPPD crystal‑driven inflammations so painful?
When uric acid or calcium pyrophosphate crystals settle in a joint, they are phagocytosed by macrophages. Inside the cell, the crystals damage lysosomes, releasing cathepsin‑B and prompting NLRP3 activation. The resulting surge of IL‑1β recruits neutrophils, which release enzymes that damage tissue and create that classic throbbing pain.
Therapies that block IL‑1β (canakinumab) or dampen NLRP3 (dapansutrile) have shown impressive results. If you want to read more about the disease mechanisms, check out this article on crystal‑driven inflammation.
What is the link between silica exposure and silicosis?
Silica particles are tiny, jagged shards that the lungs can’t easily clear. Macrophages attempt to engulf them, but the fragments damage the lysosomal membrane, releasing ROS and triggering the NLRP3 inflammasome. Over months or years, the chronic release of IL‑1β and TGF‑β fuels fibrotic scar tissue, leading to the stiff, breathing‑limiting lungs characteristic of silicosis.
Understanding this pathway has opened doors to potential anti‑inflammasome drugs, though none are yet approved for routine silicosis care. For a deeper dive into the underlying causes, see silicosis causes.
How do toxic particle diseases hijack macrophage signaling?
Asbestos, carbon nanotubes, and other occupational particles behave much like silica. They provoke ROS production, activate MAPK pathways, and keep NF‑κB turned on for years. The result? Chronic inflammation, tissue remodeling, and increased cancer risk. Workers in high‑exposure jobs are therefore monitored closely, and new protective regulations now recommend regular lung‑function testing.
What makes crystal‑driven inflammation a common thread?
All these conditions—gout, CPPD, silicosis, and various toxic particle diseases—share three critical steps:
- Phagocytosis of a rigid particle or crystal.
- Lysosomal rupture → release of danger‑associated molecular patterns (DAMPs).
- Activation of the NLRP3 inflammasome → IL‑1β & IL‑18 release.
Because the downstream events are so similar, researchers are hunting “pan‑crystal” inhibitors that could blunt the inflammasome across multiple diseases.
Targeting the Pathways
Which pathway inhibitors are already approved?
Here’s a quick rundown of drugs you might already have heard of:
- JAK inhibitors (tofacitinib, baricitinib) – approved for RA, ulcerative colitis, and even COVID‑19 cytokine storm.
- IL‑1 blockers (anakinra, canakinumab) – used in gout flares, CAPS, and some autoinflammatory syndromes.
- TNF‑α antagonists (adalimumab, etanercept) – cornerstone therapies for many inflammatory joint diseases.
- Colchicine – a centuries‑old crystal‑disruptor that also dampens NLRP3 activation.
What experimental drugs are in the pipeline?
Exciting candidates include:
- MCC950 – a selective NLRP3 inhibitor currently in Phase II trials for gout and atherosclerosis.
- STING agonists/antagonists – being evaluated for viral infections and autoimmune disease, respectively.
- PI3K‑γ inhibitors – aimed at reshaping macrophage metabolism in cancer and fibrotic lung disease.
Clinical‑trials‑at‑a‑glance
If you’re curious about the current status, a quick look at clinicaltrials.gov shows:
- 12 active studies on NLRP3 inhibitors.
- 7 trials combining JAK inhibitors with anti‑fibrotic agents for silicosis‑related lung remodeling.
- 3 Phase I studies testing STING modulators in systemic lupus.
How can we balance benefits vs. risks of pathway modulation?
Every powerful drug comes with a trade‑off. JAK blockers, for example, can increase infection risk because they blunt the very cytokine signals needed to fight microbes. IL‑1 blockers may raise the chance of neutropenia. The key is personalized medicine: measuring baseline cytokine levels, checking for latent infections, and monitoring blood counts regularly.
Think of it like tuning a radio—turn the volume up too high and you’ll get distortion; keep it just right, and the music (your health) plays beautifully.
Can lifestyle or diet influence macrophage signaling?
Good news! You don’t need a prescription to give your macrophages a little love:
- Omega‑3 fatty acids (found in salmon, flaxseed) dampen NF‑κB activation.
- Caloric restriction or intermittent fasting activates AMPK, which nudges macrophages toward an anti‑inflammatory phenotype.
- Regular exercise improves mitochondrial health, reducing ROS‑driven NLRP3 activation.
So next time you brew a cup of green tea, remember you’re also giving your innate immune system a gentle hug.
Future directions: personalized macrophage‑pathway medicine
Advances in single‑cell RNA sequencing now let researchers map the exact signaling fingerprint of a patient’s macrophages. Combine that with AI‑driven drug‑repurposing platforms, and we might soon have apps that suggest the best pathway‑targeted therapy based on a tiny blood sample.
Ethically, we must protect patient data and ensure equitable access, but the potential to transform treatment—from “one‑size‑fits‑all” to “tailored‑to‑your‑cells”—is thrilling.
Practical Evaluation Guide
- Identify the pathway. Look at your physician’s lab results—elevated CRP might hint at NF‑κB activation; high IL‑6 could point to JAK/STAT.
- Check the evidence. Search for Phase II/III trial data; ensure the drug has robust safety profiles.
- Assess safety. Ask about infection risk, liver function monitoring, or potential drug‑drug interactions.
- Match disease to pathway. Use the sections above—if you have gout, the NLRP3 inflammasome is the prime target; for RA, consider JAK or TNF‑α blockers.
- Discuss with your clinician. Bring the pathway map, ask about biomarker testing, and decide together what’s best for you.
Conclusion
Macrophage pathways are the hidden wiring that decides whether our immune system calms down or turns up the heat. By understanding the core routes—TLR/NF‑κB, JAK/STAT, NLRP3 inflammasome, and the metabolic switches—we can see why they show up in everything from painful gout attacks to stubborn lung fibrosis caused by silica. The good news is that science is already delivering drugs that specifically silence the trouble‑makers, and lifestyle tweaks can give those cells a healthier backdrop.
If any of this sparked a question—maybe you’re dealing with joint pain, or you work in an environment with dust exposure—don’t hesitate to bring it up with a health professional. Sharing what you’ve learned can be the first step toward a clearer, calmer immune system.
What’s your experience with macrophage‑related conditions? Have you tried any of the therapies mentioned? I’d love to hear your story. Together we can keep the conversation going and help each other stay healthy.



















Leave a Reply
You must be logged in to post a comment.